 Cambiar idioma
Cambiar idioma Inglés
Inglés Catalán
CatalánRetina / Retinosis Pigmentaria
¿Qué es la retinosis pigmentaria?
La retinosis pigmentaria (RP) o Retinitis Pigmentosa (RP) es una distrofia hereditaria de la retina de carácter degenerativo y progresivo. Se trata de un grupo de degeneraciones retinianas progresivas hereditarias que afecta aproximadamente a 1/4000 personas de la población general. La incidencia estimada en Estados Unidos es de 100.000 personas, con una prevalencia mundial de 1 de cada 3000 a 1 de cada 7000 habitantes.En España el número de afectados/as supera las 15.000 personas, estimándose en 500.000 las portadoras de los genes defectuosos y, por tanto, posibles transmisores de esta enfermedad.
Afecta habitualmente a los dos ojos provocando la pérdida gradual concéntrica de la visión periférica y que en muchos casos conduce a la ceguera. La retinosis pigmentaria se puede presentar sola o asociada a otras enfermedades del cuerpo humano.Aunque aún queda mucho camino por recorrer se ha avanzado notablemente en el conocimiento de los factores que intervienen en su aparición y evolución.
Esta enfermedad puede aparecer entre los 25 y 40 años de edad. Aunque también puede ocurrir que la retinosis aparezca en personas con menos de 20 años, aunque es raro que se manifieste antes de la adolescencia. También puede aparecer a partir de los 50.
Sobre el pronóstico de la retinosis pigmentaria paradójicamente suele ser cuanto más tarde menor aunque depende de la forma de herencia y de la edad de la persona afectada.
Esta enfermedad se puede presentar sola, y la denominamos “no sindrómica” o asociada a otros síndromes, denominándose en este caso “retinosis pigmentaria sindrómica”. El síndrome mas frecuente que asocia RP es el que asocia sordera, al que conocemos como Síndrome de Usher, el segundo en frecuencia es el Síndrome de Bardet-Biedl asociado a polidactilia, hipogonadismo, obesidad y retraso mental.La RP sindrómica suele ser de herencia recesiva por lo que la forma no sindrómica es la forma de presentación más frecuente.
¿Por qué es una enfermedad rara?
Fue descrita por primera vez por Donders en 1855 y desde entonces no se ha cesado en el empeño del conocimiento de los diversos factores y desordenes genéticos que intervienen en su aparición y desarrollo, quedando aun mucho recorrido por hacer.
La dificultad del entendimiento de esta enfermedad radica en la complejidad de su fisiopatología y genética, ya que hasta la fecha las mutaciones halladas tan solo explican menos de la mitad de los casos de retinosis pigmentarias.
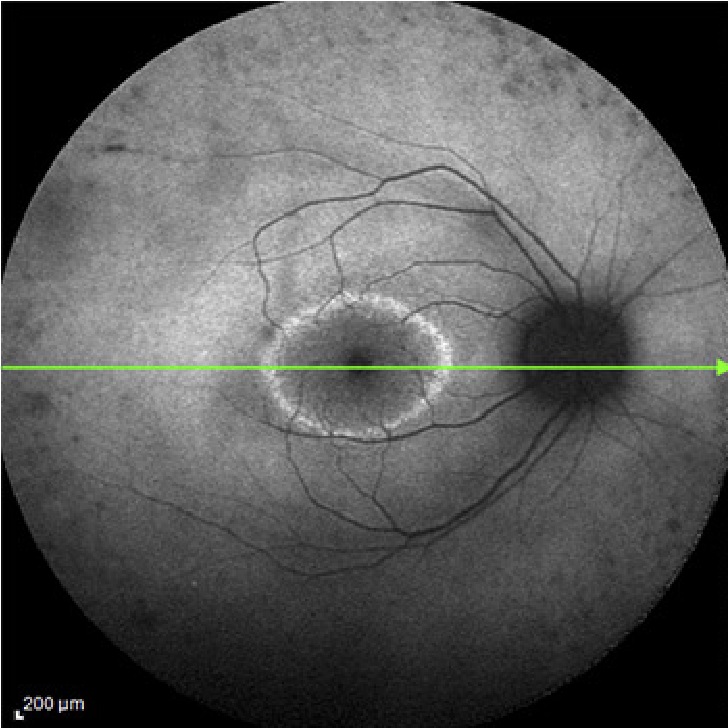
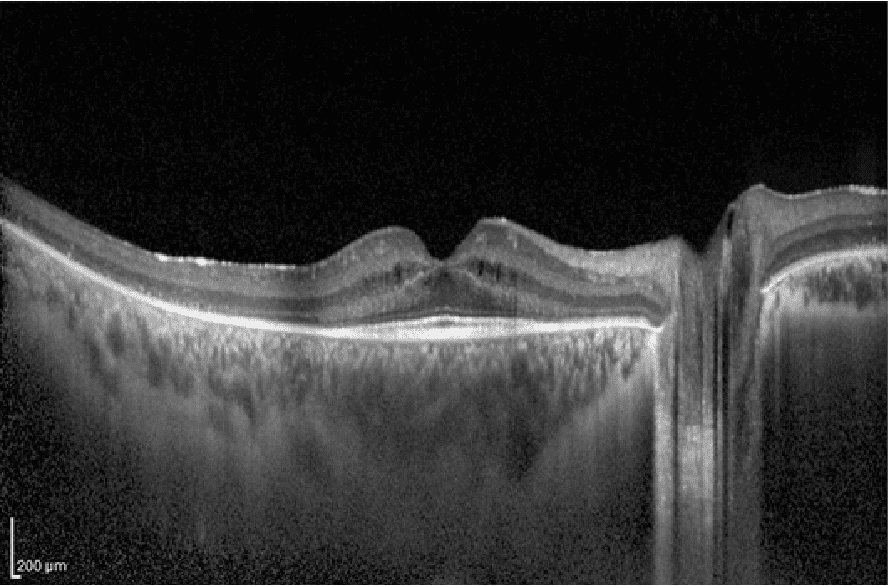
El principal problema que existe, es la gran variabilidad de mutaciones genéticas que pueden producir la misma lesión en la retina, y también al contrario, cómo la misma mutación encontrada en una familia sea capaz de expresar diferentes formas y grados de enfermedad. Esto sugiere que no solo existan factores genéticos implicados, sino también ambientales, pudiéndose modular el desarrollo de la enfermedad.
Entre los factores ambientales que pueden estar implicados se ha descrito la luteína, las vitaminas antioxidantes y dieta rica en DHA-omega3 como factores protectores, retrasando la pérdida de agudeza visual, y por el contrario, los hábitos tóxicos (tabaco, alcohol….), el estrés, la ansiedad y la exposición a la luz parecen acelerar la enfermedad, aunque no se han realizado estudios concluyentes.
Tipos de retinosis pigmentaria
Podemos diferenciar cuatro tipos de retinosis pigmentaria:
→ Patrón Autosómico Dominante (ADRP): la enfermedad la tiene uno de sus padres y alguno de sus hijos. Entre un 6% y un 15%
→ Patrón Autosómico Recesivo (ARRP): la enfermedad el padre no la tiene pero la puede trasmitir genéticamente al hijo y es él quien sufre esta patología. El deterioro comienza en la edad adulta conservando la buena visión por encima de los 60 años.Es la forma hereditaria más frecuente. Entre un 22% y un 26%.
→ Patrón ligado al cromosoma X (XLRP): son las madres las que trasmiten esta enfermedad a sus hijos varones. Entre el 1% y el 10% de los casos.
→ Patrón simple (mutaciones esporádicas aisladas sin historia familiar conocida): la persona que lo trasmite es la primera en padecer los síntomas de la retinosis pigmentaria. El 50% de los casos.
Según el estudio realizado por el Grupo Español Multicéntrico de investigación sobre retinopatías hereditarias la forma hereditaria mas frecuente en nuestro país resultó ser la mutación esporádica en un 40% de los casos, seguida de las autosómicas recesivas en un 39% y las dominantes en un 14%, siendo tan solo un 6% las asociados al cromosoma X. Esta distribución es muy diferente en otras regiones de Europa siendo por ejemplo Gran Bretaña donde más casos ligados al X o dominantes se presentan.


Causas de la retinosis pigmentaria
La causa de una retinosis pigmentaria más frecuente es genética, al menos el 50% de los casos. El patrón de herencia es clave ya que la pérdida de visión es precoz en las formas ligadas al sexo, incluso apreciable desde los 4-6 años llegando a ser ciegos legales (visión menor de 0,1) a la edad de 30-40 años. Existen numerosos genes que pueden provocarla, siendo 120 los que hasta la fecha de hoy se han identificado. No obstante, también existen factores ambientales que pueden afectar tanto en su progresión como en su detección.
Tener retinosis pigmentaria por tanto significa una degeneración y muerte celular de los fotoreceptores (células de la retina), de los bastones (responsables de la visión del campo periférico) y, en fases más avanzadas, de los conos, responsables de la visión central, llegando así a la ceguera.
Síntomas de la retinosis pigmentaria
El primer síntoma que el paciente va a experimentar es la dificultad para la visión nocturna y en condiciones de baja luminosidad. Del mismo modo, se produce una pérdida progresiva del campo visual periférico hasta formar una visión en túnel. Al ser un deterioro muy lento e indoloro el paciente puede llegar a retrasar su visita al oftalmólogo. Pueden llegar a transcurrir incluso años desde el inicio de los primeros síntomas.
Es importante señalar que esta enfermedad no afecta a todas las personas por igual, incluso aunque sean de la misma familia.
Otros síntomas asociados a la retinosis pigmentaria:
→ Defectos de la visión de colores: suele afectar más al eje azul/amarillo y se produce en estadios más avanzados.
→ Pérdida de la visión central: no ocurre en las primeras fases.
→ Deslumbramientos y fotopsias: se perciben luces en la periferia de su campo de visión, sobre todo en condiciones de excesiva lumnisidad. Esta condición obliga a usar gafas de sol con filtros especiales.
→ Distorsión de la agudeza visual por enfermedades asociadas como las cataratas, membrana epirretiniana o edema macular quístico.
Diagnóstico de la retinosis pigmentaria
Un correcto diagnóstico de la retinosis pigmentaria debe realizarlo un especialista oftalmólogo en retina. Una vez en consulta, el profesional procederá a realizar un completo examen del ojo entre los que se encuentran:

Prueba de agudeza visual

Tomografía de Coherencia Óptica (OCT)

Campo visual 24/2 y 10/2

Prueba de refracción
Prueba de determinación defectiva del color

Examen de la retina (retinografía)

Oftalmoscopia con lámpara de hendidura
Tratamiento de la retinosis pigmentaria
Actualmente están en marcha numerosos estudios y ensayos clínicos a nivel mundial que analizan diferentes estrategias terapéuticas para frenar el desarrollo de la enfermedad y aunque se está avanzando mucho, actualmente no existe ningún tratamiento eficaz para detener la evolución de la retinosis pigmentaria.
La complejidad de la base genética de esta enfermedad aun no conocida del todo dificulta el desarrollo de terapias, ahora bien los grandes avances científicos en la biología molecular hace que el futuro sea esperanzador en cuanto a posible terapias génicas o de transferencia celular de células madre, epitelio pigmentario, o fotorreceptores. También el gran avance tecnológico e informático ha llevado al desarrollo de implantes de microchips subretinianos con resultados no desdeñables que dan algo de luz y esperanza al futuro de estos enfermos.
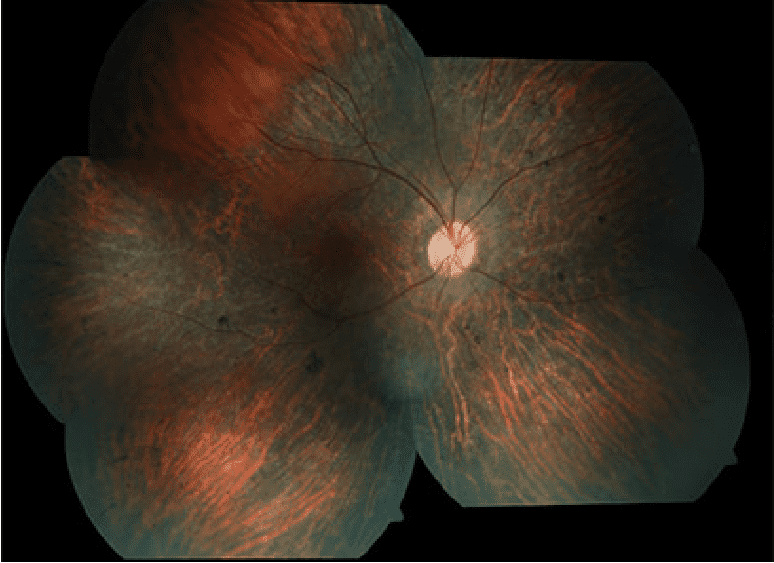
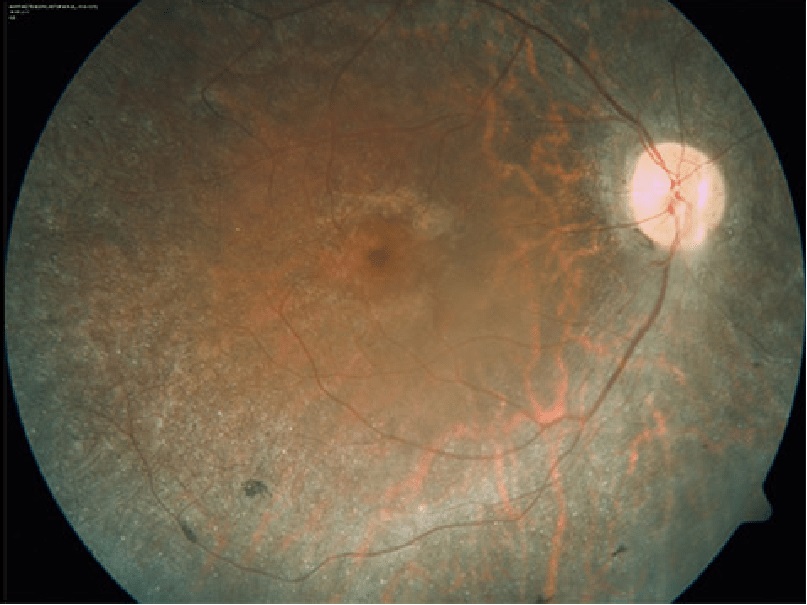
Hoy en día las terapias que disponemos se limitan a ralentizar el proceso degenerativo:
→ Protegiéndose de la luz solar.
→ Mediante vitaminas.
→ Tratando las complicaciones que con frecuencia se puede desarrollar durante la evolución de esta enfermedad: cataratas, edema macular, o membranas epirretinianas maculares.
→ Es por esto que el seguimiento con sistemas de diagnóstico de última generación son indispensables.
→ Tampoco hay que olvidar el consejo genético que deben recibir de estos enfermos.
Prevención de la retinosis pigmentaria
Un examen oftalmológico completo puede ayudar a determinar si padece retinosis pigmentaria. También lo puede ser un estudio de ADN genético pero la realidad es que hoy en día un análisis de este tipo tiene un alto coste, así como laborioso ya que requiere que haya un componente claro hereditario (al menos tres afectados), extracción de sangre para conocer el ADN y la colaboración de los familiares sanos.
Oftalmólogos especializados en retinosis pigmentaria en nuestros centros

















Preguntas frecuentes
→ ¿Qué significa que la retinosis pigmentaria es degenerativa?
Esta enfermedad es degenerativa porque la pérdida visual es progresiva con el tiempo. No se trata de algo repentino, sino gradual, de forma que la agudeza visual disminuye poco a poco.
→ ¿La retinosis pigmentaria tiene cura?
Aún no existe ninguna cura para la retinosis pigmentaria pero existen diferentes grupos de investigación y estudios en la actualidad para desarrollar diferentes opciones de tratamientos con avances muy promotedores.
→ ¿La retinosis pigmentaria puede afectar a niños?
Sí, aunque es raro que se produzca antes de los 20 años de edad. Sin embargo, sí puede darse en los niños cuando nacen con pocos o ningún fotoreceptor en la retina por lo que el primer síntoma será la pérdida de atención visual.
→ ¿Por qué la retinosis es una enfermedad genética?
Porque nacemos con esa enfermedad al producirse una mutación en nuestros genes. O, al menos, una predisposición a padecerla.
→ ¿Puede estancarse el desarrollo de la retinosis?
Por lo general suele haber periodos (incluso de años) en los que la agudeza visual y el campo varían muy poco. Pero la pérdida visual, aunque pequeña, suele ser continua.
